Effect of Intensive Atorvastatin Therapy on Coronary Atherosclerosis Progression, Composition, Arterial Remodeling, and Microvascular Function
Abstract:
Background. There is a discrepancy between the marked reduction in adverse events with statins and their modest effect on atheroma regression. We hypothesized that, in a Western population, high-dose atorvastatin will result in alterations in coronary atheroma composition, phenotype, and microvascular function.
Methods. Serial coronary radiofrequency intravascular ultrasound (VH-IVUS), coronary flow reserve (CFR), and hyperemic microvascular resistance (HMR) were performed at baseline and after 6 months of treatment with 80 mg atorvastatin in 20 patients with moderate coronary artery disease (CAD). For each VH-IVUS frame (n = 2249), changes in total plaque atheroma, composition, and phenotype (pathological intimal thickening, fibrotic plaque, fibroatheroma), and serial remodeling were assessed.
Results. Total serum cholesterol decreased from 186.0 mg/dL (interquartile range [IQR], 168.0 to 212.5 mg/dL) to 139.0 mg/dL (IQR, 124.3 to 151.3 mg/dL). Percent atheroma volume did not change significantly (-0.5% [IQR, -2.8% to 3.7%]; P=.90) and serial remodeling analysis demonstrated 40% constrictive, 24% incomplete, and 36% expansive patterns. There was a trend toward lower percent fibrous tissue (-3.47 ± 1.78%; P=.07) and percent fibro-fatty tissue (-2.52 ± 1.24%; P=.06) and increase in percent necrotic core (+2.74 ± 1.65%; P=.11) and percent dense calcium (+1.99 ± 0.81; P=.02), which translated into significantly less pathological intimal thickening (4% vs 12%; P<.0001) and more fibroatheromas (67% vs 57%; P<.0001) at follow-up compared to baseline. There were modest non-significant improvements in CFR (+0.26 [IQR, -0.37 to 0.76]; P=.23) and HMR (-0.22 [IQR, -0.56 to 0.28]; P=.12).
Conclusions. In this pilot study of Western patients with moderate CAD, high-dose atorvastatin resulted in alterations in coronary atheroma composition with corresponding changes in plaque phenotype and modest improvement in coronary microvascular function.
J INVASIVE CARDIOL 2012;24(10):522-529
Key words: atherosclerosis, coronary artery, intravascular ultrasound, microvascular function, statin, virtual histology
______________________________________________________
Contemporary therapy of atherosclerosis includes risk-factor modification and pharmacologic therapy aimed at stabilizing or regressing disease. The cornerstone of anti-atherosclerotic therapy, 5-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) are thought to exert their protective effect through a number of mechanisms including low density lipoprotein (LDL) lowering, halting progression of atherosclerosis, anti-inflammatory effects, and other pleiotropic effects.1-4 Several prospective trials have shown a correlation between the degree of LDL lowering by statins and their effects on reducing progression and/or inducing regression of atheroma in patients with coronary artery disease (CAD).5-7
Nevertheless, there remains a discrepancy between the significant relative-risk reduction in clinical events with statins and their relatively modest effect on atheroma regression. It has therefore been proposed that the protective effects of statins are mediated through alterations in coronary atheroma composition and improvement in endothelial function, serum viscosity, and microvascular function.3,8,9 Existing studies evaluating the effect of statins on atheroma composition have been limited to Asian populations, in which the more fibrotic and calcified atherosclerotic phenotype differs from that observed in Western populations.10,11
Accordingly, the aim of this pilot study was to assess the change in coronary atheroma size, composition, and phenotype, and serial remodeling using radiofrequency intravascular ultrasound (virtual histology intravascular ultrasound [VH-IVUS]), and coronary microvascular function in a Western population treated with high-dose atorvastatin (80 mg) for 6 months. We hypothesized that in a Western cohort with moderate CAD, high-dose atorvastatin will result in significant alterations in atheroma composition, plaque phenotype, and coronary microvascular function.
Method
Study population. We enrolled patients presenting to the cardiac catheterization laboratory at Emory University Hospital between December 2007 and January 2009 with an abnormal non-invasive stress test, stable angina, or stabilized acute coronary syndrome who were found to have moderate lesions requiring invasive physiologic evaluation. Exclusion criteria were myocardial infarction, cardiogenic shock or hemodynamic instability, lesion requiring percutaneous or surgical revascularization, coronary artery bypass surgery (CABG), severe valvular heart disease, presence of visual coronary collaterals, inability to provide informed consent, patients on a maximum dose of statin (atorvastatin 80 mg, simvastatin 80 mg, rosuvastatin 40 mg, pravastatin 80 mg, or fluvastatin 80 mg) or on statin treatment with an LDL ≤130, serum creatinine >1.5 mg/dL pregnancy or planned pregnancy, liver disease, INR >1.8, significant hematologic diseases, or uncontrolled diabetes requiring intensification of therapy, and uncontrolled hypertension requiring the addition of angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers.
Twenty-seven patients were enrolled in this pilot study and underwent baseline VH-IVUS and physiologic assessment. All patients underwent lipid profile assessment at baseline and 6-month follow-up, and received optimal medical therapy for cardiovascular risk factors including atorvastatin 80 mg daily. The patients were given 40 mg for 1 week and the dosage was then increased to 80 mg. At 8 weeks, the patients returned for assessment of their lipid and liver profile and renal function. Of the 27 enrolled patients, 1 patient was withdrawn because of non-compliance with high-dose atorvastatin and 6 patients did not return for follow-up assessment. Therefore, 20 patients returned to the cardiac catheterization laboratory at 6 months and underwent repeat evaluation for the assessment of atheroma progression, change in atheroma composition, serial remodeling, and change in coronary epicardial and microvascular functions. All enrolled patients provided written informed consent prior to the baseline procedure. The study was approved by the Emory University Institutional Review Board. The funders had no role in the conduct of the trial or in the analysis or interpretation of the data.
Coronary physiology assessment. Fractional flow reserve (FFR), coronary flow reserve (CFR), and hyperemic microvascular resistance (HMR) were measured as described previously12,13 using a 0.014˝ pressure and Doppler flow velocity monitoring guidewire (ComboWire, Volcano Corporation) and after infusion of 140 μg/kg/min intravenous adenosine for 3 minutes to induce maximal coronary hyperemia. Fractional flow reserve was defined as the ratio of mean distal to aortic pressure during hyperemia, CFR as the hyperemic to baseline average peak velocity, and HMR as the ratio of mean distal pressure to average peak velocity during maximal hyperemia.14,15
Gray-scale and VH-IVUS image acquisition and analysis. IVUS image acquisition was performed as described previously12,16 after administration of 200 µg intracoronary nitroglycerin using a phased-array 20 MHz Eagle Eye Gold Catheter and s5 Imaging System (Volcano Corporation). The IVUS catheter was located as distal as possible in the coronary artery of interest using a fiduciary side branch as the starting point. Automated motorized pullback (0.5 mm/s) was performed and IVUS images were continuously acquired up to the guiding catheter in the aorta. At least 60 mm of the proximal vessel was sampled in all patients. The ECG-gated gray-scale IVUS images and radiofrequency signals were acquired and stored for off-line analysis. IVUS radiofrequency data were acquired at the peak of the R-wave. Angiography was used to record the IVUS catheter starting position and its relationship to adjacent anatomic landmarks, such as diagonal or septal branches. Off-line volumetric analysis of the entire imaged segment was performed by a single experienced investigator blinded to the patients’ clinical data and to the time of acquisition of the data (eg, baseline or 6-month follow-up) according to criteria of the American College of Cardiology Clinical Consensus Document on IVUS,17 using Volcano Image Analysis Software (VIAS) V3.0 (Volcano Corporation).12,13,16,18 First, baseline and 6-month follow-up IVUS images were edited separately in a random order. Subsequently, once the editing of all images was completed, co-registration of baseline and follow-up VH-IVUS frames was done precisely by side-by-side review of images on a display and using anatomical landmarks according to the Clinical Expert Consensus Document on Standards for Acquisition, Measurement, and Reporting of Intravascular Ultrasound Regression/Progression Studies.19 The distal end of the target segment was determined by the presence of a reproducible index side branch and the proximal end was determined by the presence of aorta. Our method of frame-per-frame co-registration of baseline and follow-up VH-IVUS images has been previously reported.12 IVUS measurements of the external elastic membrane (EEM), atheroma (plaque and media: EEM – lumen), and lumen cross-sectional areas (CSA) were performed for every recorded VH-IVUS frame (0.5 mm thickness). Atheroma burden was calculated as atheroma CSA divided by EEM CSA.20 In order to assess atheroma composition, absolute and relative (percentage) CSA of VH-IVUS parameters (fibro-fatty tissue [FF], fibrous tissue [FI], necrotic core [NC], and dense calcium [DC]) were measured for each IVUS frame.21-23 For the segment-level (frame-level) analysis, change in CSA of EEM, lumen, atheroma, and four components of atheroma (FF, FI, NC, and DC), as well as atheroma burden were calculated as follow-up values minus baseline values for each IVUS segment.19 For the vessel-level analysis, volumetric IVUS measurements for each vessel were performed using the Simpsons’s Rule.17,19 Total atheroma volume was calculated as S(EEM CSA – Lumen CSA), and percent atheroma volume as S([EEM CSA – Lumen CSA])/(EEM CSA) ×100.19 Atheroma progression was defined as Δ atheroma (follow-up atheroma – baseline atheroma) CSA >0 and atheroma regression/no progression was defined as Δ atheroma CSA ≤0). Intra-observer variability of IVUS measurements from our laboratory has been previously reported.12
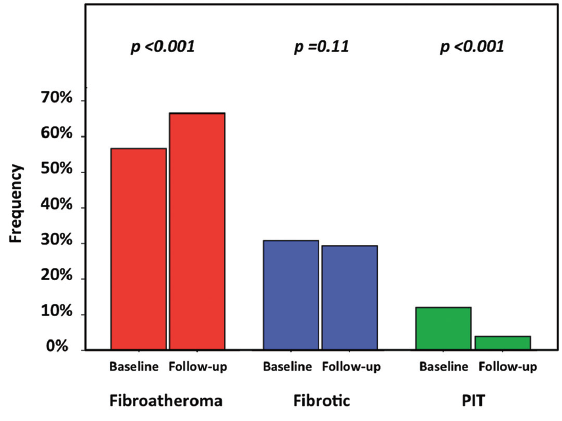
To assess plaque phenotype, coronary plaques were classified qualitatively based on atheroma composition.21,24 Pathological intimal thickening consisted primarily of FI and FF tissue with FF ≥15% with <10% confluent NC and <10% confluent DC. Fibrotic plaque consisted of mainly FI tissue with <10% confluent NC, <15% FF tissue, and <10% confluent DC. Fibroatheroma was defined as a plaque with ≥10% confluent NC.21,24
Serial remodeling was defined as expansive (positive), incomplete, and constrictive (negative).17,19 Based on the IVUS clinical expert consensus documents, the serial remodeling index of each VH-IVUS frame was calculated as ΔEEM CSA (EEM CSA at follow-up – EEM CSA at baseline). Positive ΔEEM CSA was defined as positive remodeling and negative ΔEEM CSA as constrictive remodeling. Furthermore, segments with positive remodeling were subdivided as expansive where ΔEEM CSA/Δatheroma CSA (atheroma CSA at follow-up – atheroma CSA at baseline) was >1 or incomplete where ΔEEM CSA/Δatheroma CSA was between 0 and 1.0.17,19
The pre-specified primary endpoint of this pilot study was change in plaque composition and phenotype and the secondary endpoints were serial arterial remodeling and change in microvascular function from baseline to 6-month follow-up.
Statistical analysis. Continuous variables are described as mean ± standard error (or median and interquartile range [IQR], as appropriate), and categorical variables as counts and proportions. Repeated measures analysis using mixed effects linear models was conducted to account for the correlation between multiple observations from the same subject. The model-based means are unbiased with unbalanced and missing data, so long as the missing at random can be assumed. Valid standard errors were calculated, and efficient statistical tests were performed. A compound symmetric variance-covariance form among the segment-level data was used. To determine whether a significant difference occurred from baseline to follow-up in vessel-level data, the nonparametric Wilcoxon signed rank test was used. For categorical outcomes, generalized estimating equation logistic regression models were constructed; an exchangeable correlation structure was assumed for the repeated binary response within subject. SAS version 9.2 (SAS Institute Inc) was used in the data analysis. Statistical tests were two-sided; the significance level was set at .05.
The study has been registered at clinicaltrial.gov (NCT00576576).
Results
Left anterior descending coronary arteries from 20 patients were analyzed. The baseline clinical and demographic characteristics of the study population are summarized in Table 1. The median age was 54 years (46 to 68 years) and 65% were men. From these 20 vessels, there were 2249 VH-IVUS frames (segments) available for the current analyses (120 frames per vessel [80 to 133 frames per vessel]). At baseline, the average atheroma burden was 40.62 ± 2.70%, FFR was 0.90 (0.82 to 0.96), and CFR was 2.35 (2.03 to 2.59). During 6-month therapy, the total cholesterol decreased from 186.0 mg/dL (168.0 to 212.5 mg/dL) to 139.0 mg/dL (124.3 to 151.3 mg/dL) (25% reduction; P<.0001) and the LDL cholesterol decreased from 118.5 mg/dL (105.3 to 140.5 mg/dL) to 70.5 mg/dL (54.3 to 87.5 mg/dL) (41% reduction; P<.001), but HDL (39.5 to 42.5; P=.13) and triglycerides (115.5 to 107.0; P=.21) did not demonstrate any statistically significant change (Table 2).
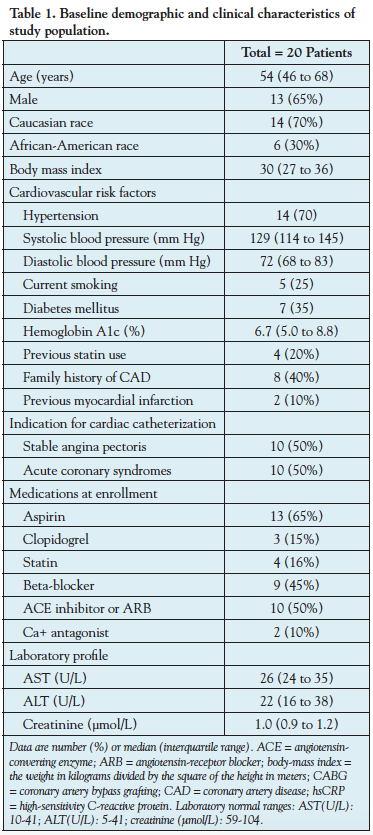
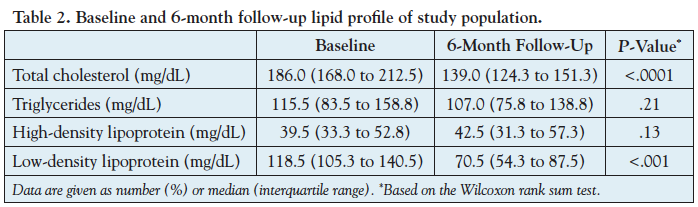
Atheroma volume regression/progression. Segment-level analyses demonstrated no significant change in atheroma CSA (-0.08 ± 0.10 mm2; P=.47) and atheroma burden (+0.01 ± 0.92%, P=.99) at 6-month follow-up compared to baseline. Vessel-level analyses demonstrated similar results with no significant reductions in atheroma volume (-4.0 mm3 [-20.4 to 11.6 mm3]; [1.3% reduction]; P=.20) and percent atheroma volume (-0.5% [-2.8 to 3.7%]; P=.90) (Table 3). Of the total 2249 IVUS frames, a total of 1022 (45%) demonstrated progression and 1227 (55%) regression/no progression of atheroma CSA.
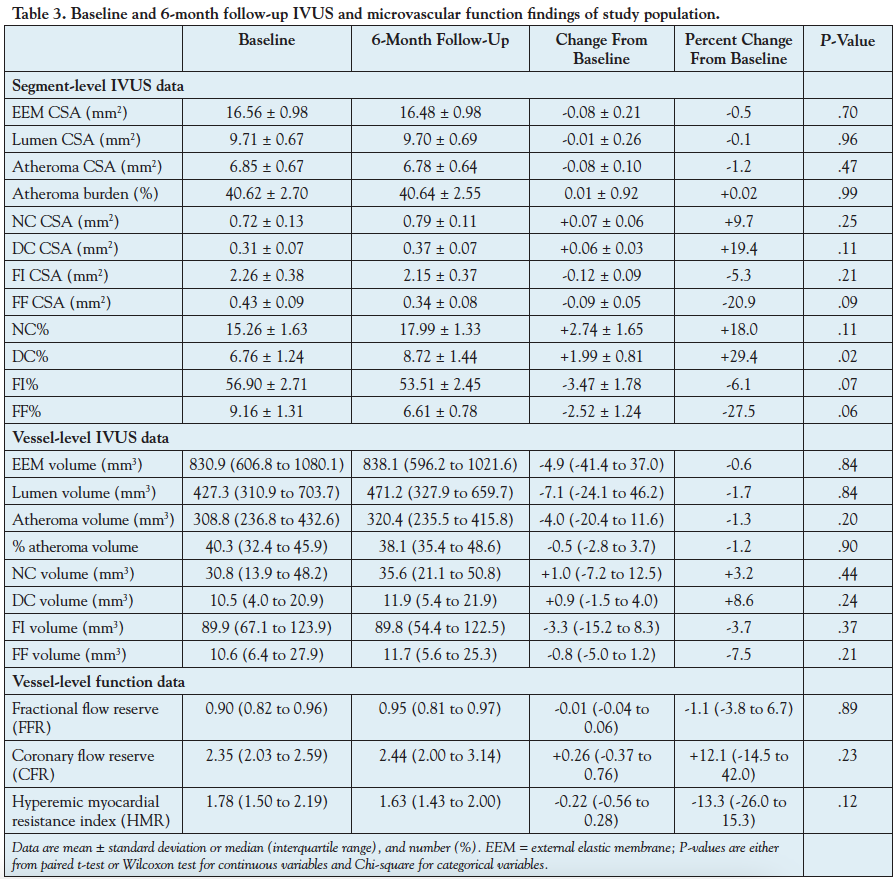
Change in plaque phenotype. Analysis of atheroma composition at the segment-level data demonstrated trends toward reductions in FI% (-3.47 ± 1.78; P=.07) and FF% (-2.52 ± 1.24; P=.06), increase in NC% (+2.74 ± 1.65; P=.11), and a significant increase in DC% (+1.99 ± 0.81; P=.02) (Table 3). Consistent with the quantitative trends observed in changes in atheroma composition, plaque phenotype pattern showed a significant change at 6-month follow-up compared to baseline, with significantly less pathological intimal thickening (4% vs 12%; P<.0001) and more fibroatheromas (67% vs 57%; P<.0001), but equal number of fibrotic plaques (29% vs 31%; P=.11) (Figure 1). At the vessel-level analysis, these trends did not hold, with no significant change in FI volume (-3.3 mm3 [-15.2 to 8.3 mm3]; P=.37), FF volume (-0.8 mm3 [-5.0 to 1.2 mm3]; P=.21), NC volume (+1.0 mm3 [-7.2 to 12.5 mm3]; P=.44), or DC volume (+0.9 mm3 [-1.5 to 4.0 mm3]; P=.24).
Serial arterial remodeling. Compared to baseline, EEM CSA (-0.08 ± 0.21 mm2; P=.70) demonstrated no significant change. Serial remodeling analysis demonstrated 40% constrictive, 24% incomplete, and 36% expansive patterns. Interestingly, segments with atheroma progression developed 45% constrictive, 24% incomplete, and 31% expansive remodeling, whereas those with regression/no progression developed 35% constrictive, 26% incomplete, and 40% expansive remodeling.
Coronary microvascular function. Compared to baseline, CFR increased slightly (+0.26 [-0.37 to 0.76]; P=.23) and HMR decreased slightly (-0.22 [-0.56 to 0.28]; P=.12) at 6-month follow-up, suggesting a slight improvement in microvascular function, though the changes were statistically non-significant in this sample size (Table 3).
Discussion
In this pilot study, we investigated for the first time the effects of 6-month therapy with high-dose statin on changes in atheroma composition and phenotype and microvascular function in a Western population of patients with moderate CAD. We observed: (1) a halt in progression of total atheroma volume; (2) a trend toward reduction in FI and FF, increase in NC, and a significant increase in DC with a corresponding decrease in the number of pathological intimal thickening and an increase in the number of fibroatheromas; (3) a trend in the direction of improvement in coronary microvascular resistance as assessed by both CFR and HMR, although this was not statistically significant.
Statin and coronary plaque regression. Several investigations have assessed the effect of statin therapy on coronary atheroma progression/regression and composition. The finding of total atheroma regression (or no progression) in the current investigation is in line with several previous studies,5,6,25-30 specifically the studies in which high-dose atorvastatin was used.6,29,30 For the comparison with the finding of the current investigation, Table 4 summarizes selected prior investigations of atheroma progression/regression and change in atheroma composition in response to statins. In the Reversal of Atherosclerosis With Aggressive Lipid Lowering (REVERSAL) trial, patients treated with atorvastatin 80 mg daily had lower progression of coronary atherosclerosis compared with those treated with pravastatin 40 mg over 18 months with a median change in percent atheroma volume of 0.2% vs 1.6% (P<.001), respectively, similar to patients treated with atorvastatin 80 mg daily in our study (-0.5%).6 In A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (ASTEROID) trial, patients treated with rosuvastatin 40 mg daily for 24 months showed a median decrease of -0.8% in percent atheroma volume compared with baseline (P<.001).5 Moreover, in the large, multicenter Effect of Rosuvastatin versus Atorvastatin (SATURN) trial, 24 months of treatment with atorvastatin 80 mg daily resulted in 0.99% reduction in percent atheroma volume and 4.4 mm3 reduction in total atheroma volume, similar to our findings (Δ percent atheroma volume: -0.5% and Δ total atheroma volume: -4.0 mm3).29 The very similar quantitative changes in atheroma volume observed in our study and in SATURN, despite the differing statin treatment duration (6 months vs 24 months), suggest that the effect of statins on atheroma regression likely occurs within the first 6 months of therapy.
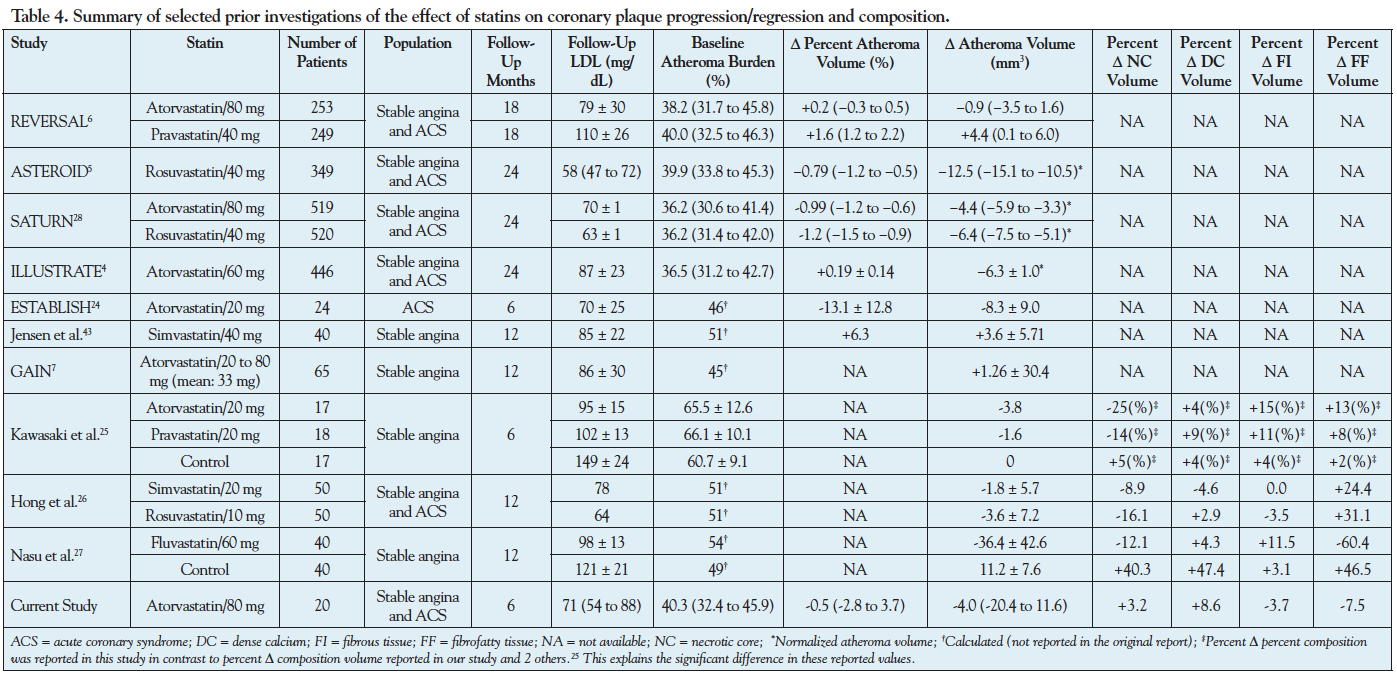
Statins and change in coronary plaque composition and phenotype. The discrepancy between the large relative-risk reduction in clinical events with statins4,9,31,32 and their relatively modest effect on atheroma regression has led to the hypothesis that in addition to systemic pleiotropic effects, statins exert their cardio-protective effect by favorably altering coronary atheroma composition. Using integrated backscatter IVUS, Kawazaki et al demonstrated a reduction in NC volume and an increase in FI volume over 6 months in Japanese patients with stable angina treated with 20 mg daily of atorvastatin or pravastatin (Table 4).25 Another study evaluated the effects of treatment with fluvastatin 60 mg daily on VH-IVUS atheroma composition of non-culprit lesions in a Japanese cohort and found reduction in FF volume, increase in FI volume, and a non-significant change in NC or DC volume.28 A third study in Korean patients reported reduction in NC volume, increase in FF volume, and a non-significant change in FI or DC volume with moderate-dose statin therapy.27
This study demonstrated a trend toward reduction in FI and FF, increase in NC, and a significant increase in DC after 6-month therapy with 80 mg atorvastatin, displaying some similarities and some differences with the aforementioned statin atheroma composition studies. Our findings of reduction in FF and FI tissue at follow-up are in keeping with observations by Nasu et al28 and Hong et al,27 respectively. Some important differences in study population and design may explain discrepant findings. In contrast to previous studies in Asian populations, our study population consisted of Caucasian and African-American patients who are known to have different atherosclerotic phenotypes.10,11 In addition, the proportion of patients with stable versus acute coronary syndromes, statin type and dose, and duration of statin therapy and follow-up vary between our study and previous studies (Table 4).
The increase in DC that we observed was also described by Kawasaki et al25 and Hong et al,27 and may be the sign of plaque stabilization. In fact, vessel calcification begins early in the process of atherosclerotic plaque development, but it is often considered a sign of advanced plaque development. This may be due to the inability of our imaging modalities to detect microcalcifications and the fact that frequently plaque ruptures are followed by intraplaque hemorrhage and calcification during the repair process. As a consequence, calcification is often seen as a sign of plaque stability,33 although the reverse may also be true. Statins have been shown to have a paradoxical effect on plaque calcification by stimulating calcification upon interaction with different types of vascular cells.34 Randomized clinical trials employing computed tomography imaging have not demonstrated slowing of progression of coronary artery calcium with statin therapy.35
We observed a slight non-statistically significant increase in NC at follow-up in this study. At first glance, this apparent increase in NC in patients treated with high-dose statin seems surprising. However, there may be a number of explanations for this observation. First, the phenotype and natural history of coronary atherosclerosis in our patient population from the southeast region of the United States (including 30% African Americans) may be different and perhaps more aggressive than that of the Asian population previously reported, rendering a halting or minimal progression of NC an improvement over a placebo or lower-dose statin therapy arm that may have shown a much larger progression of NC. This concept is highlighted in the REVERSAL trial, where more intensive statin therapy with atorvastatin 80 mg was shown to result in less progression, and not a regression, of atherosclerosis compared to pravastatin 40 mg.6 Second, baseline atheroma burden and composition may significantly impact relative changes in atheroma volume and composition, with larger atheromas being more likely to demonstrate significant relative changes than smaller atheromas. Our patient population had smaller atheromas (mean atheroma burden of 40% in our study vs 51% to 66% in other studies) and less baseline NC (mean NC of 15% in our study vs 8%, 26%, and 30% in other studies) than the previous reported studies in Asian populations. Interestingly, a recent study demonstrated that patients with smaller baseline amounts of NC or stable coronary disease tended to show progression of NC, while the patients with larger amounts of NC or acute coronary syndromes tended to show regression of NC at 12-month follow-up.36 Indeed, we have recently shown that regional change in NC is very heterogeneous and relates to local wall shear stress,12 implying that observed average changes in a vessel may be driven by regional baseline atheroma burden, content, and wall shear stress.
Statins and serial remodeling. Constrictive remodeling, a sign of plaque stabilization, has been demonstrated as the predominant response during the plaque stabilization process in patients treated with statins.7,37 Similarly, we found a greater proportion of segments with serial constrictive remodeling than with either incomplete or expansive remodeling in the total population (40% vs 24% vs 36%, respectively). Interestingly, when only segments with progression were evaluated, we observed that they are even more likely to show constrictive remodeling (45%) than other remodeling patterns (24% and 31%), as observed in a previous study.7 It has been argued that a shift of the remodeling pattern toward constrictive or negative remodeling may be a mechanism by which statins contribute to plaque stabilization even among plaques that progress over time.38-40
Statins and improvement in microvascular function. Impaired coronary microvascular function is associated with increased risk of major adverse cardiac events even in the absence of hemodynamically significant CAD.14 We found a modest improvement in microvascular function with high-dose statin therapy, evidenced by both a non-significant increase in CFR and decrease in HMR, suggesting that improvement in microvascular function, possibly through amelioration of endothelial function3 and wall shear stress10,41 and reduction in blood viscosity,8 may be another mechanism by which statins confer prognostic benefit beyond atheroma regression. In keeping with this hypothesis, we have recently shown in a cross-sectional study that statin use is an independent predictor of higher CFR in patients with non-obstructive CAD.11 This hypothesis will need to be tested in larger studies.
Study limitations. Several limitations require consideration when interpreting the results of the present pilot study. First, the current study is relatively small, has 6-month follow-up, and lacks a placebo arm due to ethical concerns of withholding statin therapy in patients with established CAD. The novelty of this study is that it is the first investigation of the effects of statin therapy on coronary atheroma composition, plaque phenotype, arterial remodeling, and microvascular function in a Western population. Another unique feature of the present study is both segment- and vessel-level IVUS analysis of the effect of statins on coronary atherosclerosis. The importance of segment-level analysis relates to the potentially heterogeneous impact of statin therapy on regional coronary plaque which may be overlooked by vessel-level analysis. The second limitation is the correlated error introduced in segment-level analysis by clustering of numerous arterial segments within patients. However, appropriate statistical methods were used to adjust for correlated error. Third, atheroma composition data were derived from VH-IVUS. While VH-IVUS has well-documented limitations for assessing atheroma composition, several studies have found its predictive accuracy to be 87% to 97%.22,23,42 In addition, clinical investigations demonstrated an association between high-risk clinical settings and larger VH-IVUS-defined necrotic core,43,44 and the recent large clinical PROSPECT trial has linked findings of VH-IVUS in conjunction with gray-scale IVUS with adverse clinical outcomes.45 Nevertheless, the findings of this pilot study need to be confirmed in large, randomized trials.
Conclusion
In this pilot study of Western patients with moderate coronary artery disease, 6-month therapy with high-dose atorvastatin resulted in alterations in coronary atheroma composition with corresponding changes in plaque phenotype and modest non-significant improvement in coronary microvascular function.
Acknowledgment. We acknowledge the Emory interventional cardiology fellows, catheterization laboratory staff, and Andrew R. King for participation in study performance.
References
- Blum A, Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation, and thrombogenesis. Atherosclerosis. 2009;203(2):325-330.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, c-reactive protein, and coronary artery disease. N Engl J Med. 2005;352(1):29-38.
- Reriani MK, Dunlay SM, Gupta B, et al. Effects of statins on coronary and peripheral endothelial function in humans: a systematic review and meta-analysis of randomized controlled trials. Eur J Cardiovasc Prev Rehabil. 2011;18(5):704-716.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the asteroid trial. JAMA. 2006;295(13):1556-1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291(9):1071-1080.
- Schartl M, Bocksch W, Koschyk DH, et al. Use of intravascular ultrasound to compare effects of different strategies of lipid-lowering therapy on plaque volume and composition in patients with coronary artery disease. Circulation. 2001;104(4):387-392.
- Rosenson RS, Tangney CC. Antiatherothrombotic properties of statins: implications for cardiovascular event reduction. JAMA. 1998;279(20):1643-1650.
- Samady H, McDaniel MC. Can statins alter coronary plaque composition assessed by radiofrequency backscatter intravascular ultrasound? JACC Cardiovasc Interv. 2009;2(7):697-700.
- Bild DE, Detrano R, Peterson D, et al. Ethnic differences in coronary calcification: the multi-ethnic study of atherosclerosis (MESA). Circulation. 2005;111(10):1313-1320.
- Budoff MJ, Nasir K, Mao S, et al. Ethnic differences of the presence and severity of coronary atherosclerosis. Atherosclerosis. 2006;187(2):343-350.
- Samady H, Eshtehardi P, McDaniel MC, et al. Coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation. 2011;124(7):779-788.
- Dhawan SS, Eshtehardi P, McDaniel MC, et al. The role of plasma aminothiols in the prediction of coronary microvascular dysfunction and plaque vulnerability. Atherosclerosis. 2011;219(1):266-272.
- Kern MJ, Samady H. Current concepts of integrated coronary physiology in the catheterization laboratory. J Am Coll Cardiol. 2010;55(3):173-185.
- Eshtehardi P, Luke J, McDaniel MC, Samady H. Intravascular imaging tools in the cardiac catheterization laboratory: comprehensive assessment of anatomy and physiology. J Cardiovasc Transl Res. 2011;4(4):393-403.
- Eshtehardi P, McDaniel MC, Suo J, et al. Association of coronary wall shear stress with atherosclerotic plaque burden, composition, and distribution in patients with coronary artery disease. J Am Heart Assoc. 2012;1(4):E002543.
- Mintz GS, Nissen SE, Anderson WD, et al. American college of cardiology clinical expert consensus document on standards for acquisition, measurement and reporting of intravascular ultrasound studies (IVUS). A report of the american college of cardiology task force on clinical expert consensus documents. J Am Coll Cardiol. 2001;37(5):1478-1492.
- Pu J, Mintz GS, Brilakis ES, et al. In vivo characterization of coronary plaques: novel findings from comparing greyscale and virtual histology intravascular ultrasound and near-infrared spectroscopy. Eur Heart J. 2012;33(3):372-383.
- Mintz GS, Garcia-Garcia HM, Nicholls SJ, et al. Clinical expert consensus document on standards for acquisition, measurement and reporting of intravascular ultrasound regression/progression studies. EuroIntervention. 2011;6(9):1123-1130, 1129.
- McDaniel MC, Eshtehardi P, Sawaya FJ, et al. Contemporary clinical applications of coronary intravascular ultrasound. JACC Cardiovasc Interv. 2011;4(11):1155-1167.
- Garcia-Garcia H, Mintz G, Lerman A, et al. Tissue characterisation using intravascular radiofrequency data analysis: recommendations for acquisition, analysis, interpretation and reporting. EuroIntervention. 2009;5(2):177-189.
- Nair A, Margolis MP, Kuban BD, Vince DG. Automated coronary plaque characterisation with intravascular ultrasound backscatter: ex vivo validation. EuroIntervention. 2007;3(1):113-120.
- Nasu K, Tsuchikane E, Katoh O, et al. Accuracy of in vivo coronary plaque morphology assessment: a validation study of in vivo virtual histology compared with in vitro histopathology. J Am Coll Cardiol. 2006;47(12):2405-2412.
- Kubo T, Maehara A, Mintz GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55(15):1590-1597.
- Kawasaki M, Sano K, Okubo M, et al. Volumetric quantitative analysis of tissue characteristics of coronary plaques after statin therapy using three-dimensional integrated backscatter intravascular ultrasound. J Am Coll Cardiol. 2005;45(12):1946-1953.
- Okazaki S, Yokoyama T, Miyauchi K, et al. Early statin treatment in patients with acute coronary syndrome: demonstration of the beneficial effect on atherosclerotic lesions by serial volumetric intravascular ultrasound analysis during half a year after coronary event: the ESTABLISH study. Circulation. 2004;110(9):1061-1068.
- Hong MK, Park DW, Lee CW, et al. Effects of statin treatments on coronary plaques assessed by volumetric virtual histology intravascular ultrasound analysis. JACC Cardiovasc Interv. 2009;2(7):679-688.
- Nasu K, Tsuchikane E, Katoh O, et al. Effect of fluvastatin on progression of coronary atherosclerotic plaque evaluated by virtual histology intravascular ultrasound. JACC Cardiovasc Interv. 2009;2(7):689-696.
- Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078-2087.
- Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356(13):1304-1316.
- Nissen SE. Effect of intensive lipid lowering on progression of coronary atherosclerosis: evidence for an early benefit from the reversal of atherosclerosis with aggressive lipid lowering (REVERSAL) trial. Am J Cardiol. 2005;96(5A):61F-68F.
- Hartmann M, Huisman J, Bose D, et al. Serial intravascular ultrasound assessment of changes in coronary atherosclerotic plaque dimensions and composition: an update. Eur J Echocardiogr. 2011;12(4):313-321.
- Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92(3):657-671.
- Wu B, Elmariah S, Kaplan FS, et al. Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts: implications for end-stage valvular heart disease. Arterioscler Thromb Vasc Biol. 2005;25(3):592-597.
- Raggi P, Davidson M, Callister TQ, et al. Aggressive versus moderate lipid-lowering therapy in hypercholesterolemic postmenopausal women: beyond endorsed lipid lowering with EBT scanning (BELLES). Circulation. 2005;112(4):563-571.
- Garcia-Garcia HM, Klauss V, Gonzalo N, et al. Relationship between cardiovascular risk factors and biomarkers with necrotic core and atheroma size: a serial intravascular ultrasound radiofrequency data analysis. Int J Cardiovasc Imaging. 2012;28(4):695-703.
- Schoenhagen P, Tuzcu EM, Apperson-Hansen C, et al. Determinants of arterial wall remodeling during lipid-lowering therapy: serial intravascular ultrasound observations from the reversal of atherosclerosis with aggressive lipid lowering therapy (REVERSAL) trial. Circulation. 2006;113(24):2826-2834.
- Hartmann M, von Birgelen C, Mintz GS, et al. Relation between baseline plaque burden and subsequent remodelling of atherosclerotic left main coronary arteries: a serial intravascular ultrasound study with long-term (> or =12 months) follow-up. Eur Heart J. 2006;27(15):1778-1784.
- Sipahi I, Tuzcu EM, Schoenhagen P, et al. Compensatory enlargement of human coronary arteries during progression of atherosclerosis is unrelated to atheroma burden: serial intravascular ultrasound observations from the REVERSAL trial. Eur Heart J. 2006;27(14):1664-1670.
- Von Birgelen C, Hartmann M, Mintz GS, et al. Spectrum of remodeling behavior observed with serial long-term (>/=12 months) follow-up intravascular ultrasound studies in left main coronary arteries. Am J Cardiol. 2004;93(9):1107-1113.
- Box FM, van der Grond J, de Craen AJ, et al. Pravastatin decreases wall shear stress and blood velocity in the internal carotid artery without affecting flow volume: results from the prosper mri study. Stroke. 2007;38(4):1374-1376.
- Nair A, Kuban BD, Tuzcu EM, et al. Coronary plaque classification with intravascular ultrasound radiofrequency data analysis. Circulation. 2002;106(17):2200-2206.
- Pundziute G, Schuijf JD, Jukema JW, et al. Evaluation of plaque characteristics in acute coronary syndromes: non-invasive assessment with multi-slice computed tomography and invasive evaluation with intravascular ultrasound radiofrequency data analysis. Eur Heart J. 2008;29(19):2373-2381.
- Rodriguez-Granillo GA, McFadden EP, Valgimigli M, et al. Coronary plaque composition of nonculprit lesions, assessed by in vivo intracoronary ultrasound radio frequency data analysis, is related to clinical presentation. Am Heart J. 2006;151(5):1020-1024.
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226-235.
- Jensen LO, Thayssen P, Pedersen KE, et al. Regression of coronary atherosclerosis by simvastatin: A serial intravascular ultrasound study. Circulation. 2004;110(3):265-270.
______________________________________________________
From the aDivision of Cardiology, Department of Medicine, Emory University School of Medicine, Atlanta, Georgia and bDepartment of Biostatistics and Bioinformatics, Emory University Rollins School of Public Health, Atlanta, Georgia.
ClinicalTrials.gov Identifier: NCT00576576.
Funding: This study was funded by Pfizer Pharmaceuticals and Volcano Corporation.
Disclosure: The authors have completed and returned the ICMJE Form for Disclosure of Potential Conflicts of Interest. The authors report no conflicts of interest regarding the content herein.
Manuscript submitted April 17, 2012, provisional acceptance given May 22, 2012, final version accepted June 19, 2012.
Address for correspondence: Habib Samady, MD, FACC, FSCAI, Professor of Medicine, Director, Interventional Cardiology, Emory University School of Medicine, 1365 Clifton Road, Suite F606, Atlanta, GA 30322. Email: hsamady@emory.edu