HIV Disease in Thrombocardiology
HIV Disease
Human immunodeficiency virus (HIV) infection results from one of two similar retroviruses (HIV-1 and HIV-2) that destroy CD4+ lymphocytes and impair cell-mediated immunity, affecting multiple organ systems, increasing the risk of certain infections and cancers. Its manifestation ranges from asymptomatic carriage to the acquired immune deficiency syndrome (AIDS), which is defined by serious opportunistic infections or cancers. Since its global epidemic in the late 1970s, and the recognition of AIDS in 1981, more than 40 million people have been infected (one-third have died, two-thirds are living with infection) with HIV disease. Approximately 90% of patients live in the developing world, 70% in Africa, and most of the new infections occur in Asia and sub-Saharan Africa. The Centers for Disease Control and Prevention (CDC) estimates that there are 650,000–900,000 people living with this disease in the United States. Public awareness of HIV has made an impact on the marked decline in the incidence of this infection among homosexual and bisexual males, but the decline is not as large as for heterosexual males, intravenous drug users and women.1
HIV attaches to and penetrates host T-cells via CD4+ molecules and chemokine receptors. After attachment, HIV RNA and enzymes (reverse transcriptase including RNA dependent DNA polymerase) are released into the cell and produce proviral DNA, which subsequently integrates into the host’s DNA with the help of HIV integrase. This is duplicated with each further cell division along with the host DNA. Proviral HIV DNA is also transcribed to viral DNA and translated into viral proteins (including envelope glycoproteins) that are assembled into HIV virions at the inner cell membrane and budded from the cell surface, producing thousands of virions. Protease, another HIV enzyme, cleaves viral proteins after budding, converting the virion into an infectious form. Thus, the main damage of HIV infection is on cell-mediated immunity, whereas humoral immunity is influenced to a lesser extent. Infected CD4+ cells produce the majority of plasma virions. After 2–8 weeks of acquisition of infection (generally no more than 12 weeks), initial Ig M response is followed by Ig G response to p24 and gp120 antigens as viremia declines.2 The diagnosis is generally made by the ELISA (enzyme-linked immunosorbent assay) assay followed by the Western blot test.
Highly active antiretroviral therapy (HAART) aims to suppress viral replication. There are currently 5 classes of medications, 3 of which inhibit reverse transcriptase blocking RNA- and DNA-dependent DNA polymerase activity.3 These are:
1. Nucleoside reverse transcriptase inhibitors: These are phosphorylated to active metabolites, which then compete for incorporation into viral DNA, resulting in inhibition of HIV reverse transcriptase enzyme competitively and terminal synthesis of the DNA chain.
2. Nucleotide reverse transcriptase inhibitors: Similar to the above group, these inhibit the reverse transcriptase without requiring phosphorylation.
3. Non-nucleoside reverse transcriptase inhibitors: Bind to reverse transcriptase enzyme directly.
4. Protease inhibitors: Inhibit viral protease enzyme that is crucial to maturation of HIV variants following budding from host cells.
5. Fusion inhibitors: Block binding of HIV to CD4+ lymphocyte receptors, which is required for HIV to enter the cells.
HIV Disease and the Heart in Thrombocardiology
HIV infection affects multiple organ systems, including cardiovascular disease. HIV cardiomyopathy secondary to direct viral damage to cardiac myocytes, which results in heart failure symptoms, is the major effect on the heart. Apart from this, the pericardial disease and endocarditis have been reported. The prevalence of cardiac morbidity and mortality was noted to be 6–7% and 1–5%, respectively.4,5 The vascular system is also affected secondary to hematologic problems. HIV may also produce cytopenias in all three bone marrow cell lines.
Prior to the availability of protease inhibitors, autopsy reports were first to describe an association between HIV and coronary artery disease. Among these, Joshi et al reported an autopsy series of 6 children with HIV who had coronary artery pathology/arteriopathy involving inflammation of endothelium leading to luminal narrowing.6 One of the children was also noted to have right coronary aneurysm with thrombus leading to myocardial infarction. Paton et al examined 8 HIV patients postmortem, 6 of whom revealed major proximal coronary atherosclerotic disease. Four of the 6 had 80–90% obstruction of the arterial lumen. It is important to note that the mean age was 27 years (range 23–32) in these subjects who had this degree of obstructive disease in the absence of traditionally known coronary artery risk factors.7 Endothelial dysfunction, manifesting itself indirectly with higher levels of vWF, tPA, beta-2 microglobulin, and soluble thrombomodulin can be seen in HIV-infected patients along with hypertriglyceridemia, all of which are mechanisms that predispose patients to atherosclerotic heart disease.
Recent literature contains reports of thrombotic episodes (deep venous thrombosis, pulmonary embolism, portal and renal vein thrombosis, etc.) occurring in HIV patients, with the incidence of thromboembolic complications being reported in the range of 0.26–7.6%; a higher incidence is seen in patients with a low CD4+ cell count, opportunistic infections, malignancy or AIDS. Among these patients, protease inhibitors are particularly implicated in this increased thromboembolic risk.8–11 In a study by Majluf-Cruz et al, the mean time from HIV infection to thrombosis was 40.5 months, with the thrombosis incidence being 1.52% (cumulative 0.30% per year) while on protease inhibitor therapy, whereas it was 0.33% (cumulative 0.055% per year) before the protease inhibitor era (p < 0.001). In their series, due to high recurrent thrombotic incidences and bleeding complications secondary to oral anticoagulants, acetylsalycilic acid was given to the patients, resulting in no subsequent events.12 Furthermore, various abnormalities have accounted for the observed hypercoagulability in HIV-infected patients, including the presence of antiphospholipid antibodies/lupus anticoagulant, hyperhomocysteinemia, elevated Factor VIII coagulant activity, decreased levels of natural anticoagulants/heparin cofactor II/antithrombin, increased level of plasminogen activator inhibitor (PAI-1), vWF and d-dimer, activated protein C resistance and increased platelet activation.13 These abnormalities correlated with the severity of HIV-related immunosuppression (HIV load), as measured by CD4+ cell counts and the presence of concurrent infectious/neoplastic diseases.
Recent studies also emphasized the increased incidence of myocardial infarction (MI), in addition to thromboembolic diseases in the HIV-infected population, especially after the introduction of HAART therapy containing protease inhibitors. Severe premature coronary artery disease has also been reported among HIV patients taking protease inhibitors.14,15 Newer risk factors such as insulin resistance, hypercholesterolemia, fat redistribution syndrome may exacerbate underlying atherosclerotic risk in patients using these medications.16 In order to identify risk factors associated with this condition, the DAD study group has looked into the relationship between myocardial infarction and the use of combination antiretroviral therapy in HIV-infected patients that included either a protease inhibitor or nonnucleoside reverse transcriptase inhibitors. Their study revealed that the combination antiretroviral therapy was independently associated with a 26% relative increase in the rate of MI per year of exposure during the first 4–6 years of their use. Other factors significantly associated with MI were older age, current or former smoking, previous cardiovascular disease, male gender, higher total cholesterol/triglyceride levels and the presence of diabetes. However, the absolute risk of MI was low, and therefore, authors concluded that the risk must be balanced against the marked benefit from antiretroviral therapy.17
Case Review
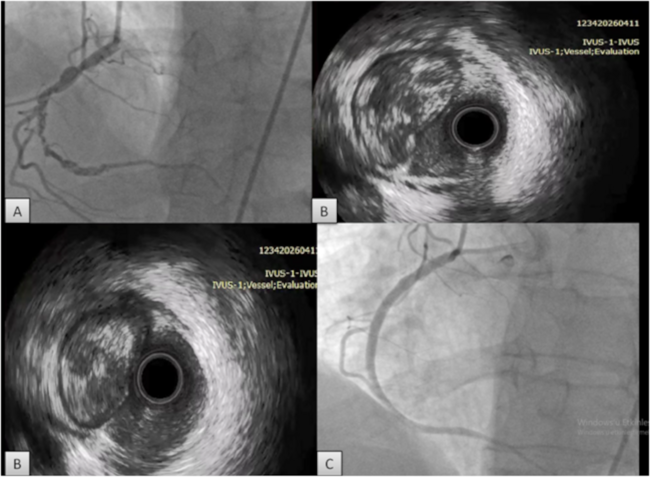
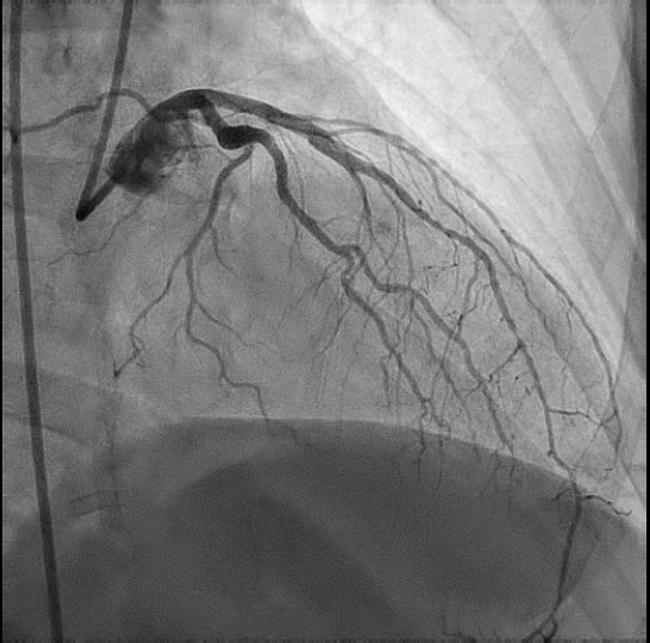
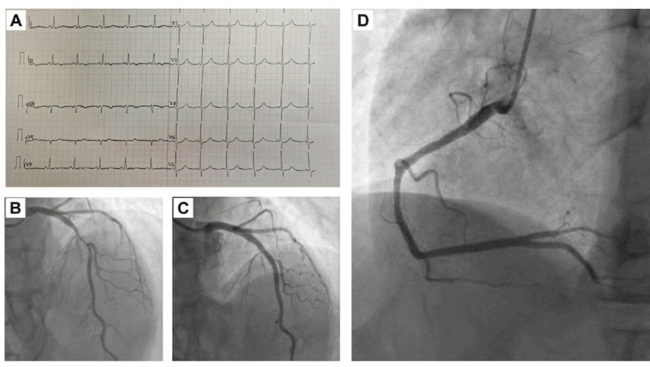
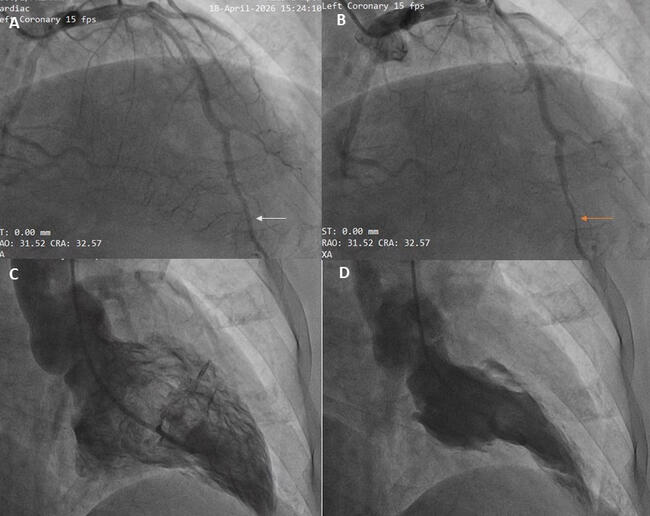
We report on a 64-year-old Hispanic female with a history of noninsulin-dependent diabetes mellitus and HIV infection who was on HAART therapy containing protease inhibitor (CD4 cell count = 670). The patient was found to have non-ST-elevation MI (NSTEMI) and underwent coronary angiography on the second day of her diagnosis. Diagnostic angiography showed 50% mid LAD (left anterior descending artery), 90% proximal LCX (left circumflex artery), 95% mid LCX, and 90% ostial RCA (right coronary artery) stenoses. After initial percutaneous transluminal coronary angioplasty (PTCA) to both proximal and mid LCX lesions, stenting of mid LCX lesion was performed using a Taxus® 2.5 x 12 mm stent (Boston Scientific Corp., Natick, Massachusetts) deployed at 10 atm, which was delivered from the mid LCX extending into the OM2 (obtuse marginal 2). Another Taxus 2.5 x 16 mm stent was deployed at the proximal LCX stenosis at 16 atm. The patient received 600 mg of clopidogrel and bivalirudin during the procedure as anticoagulant therapy. Her procedural activated clotting time (ACT) was 353 seconds. The patient complained of chest pain 1 hour postprocedurally, and was treated medically since she did not have significant electrocardiographic (ECG) changes. On the third day of hospitalization, when she was brought back for staged RCA intervention, the patient was found to have stent thrombosis of the mid LCX/OM2 stent.
Discussion
Dr. Dotter was the first to propose using an endovascular prosthesis to seal dissections and to overcome recoil.18 In 1987, Dr. Sigwart reported his initial experience with the first human stent implantation in coronary arteries.19 Initial studies from Sigwart and Serruys et al showed that thrombosis rates in coronary stenting were as much as 15–20%.19,20 Later on, the BENESTENT and STRESS trials were the first to establish the efficacy of coronary stenting in patients undergoing percutaneous coronary intervention (PCI) in 1994.21,22 With the use of intravenous heparin, warfarin, dipirydamole, low-molecular dextran and aspirin, stent thrombosis rates have been further reduced to the 5–10% range.23 These aggressive anticoagulation regimens were also associated with a high incidence of bleeding, vascular complications, prolonged hospital stays and increased cost, which offset the expected benefit of stenting.23,24 Colombo et al further demonstrated the importance of adequate stent expansion under high pressure using intravascular ultrasound (IVUS) guidance without the need for aggressive anticoagulation.25 Further studies also confirmed that combination antiplatelet therapy is superior to anticoagulant therapy in terms of reducing major adverse cardiac events (MACE).26–28 In a study by Schomig et al comparing antiplatelet versus anticoagulant therapy, they reported the occurrence of stented vessel occlusion at 0.8% in the antiplatelet therapy group, and 5.4% in the anticoagulant group.29
In most literature, factors associated with acute/subacute stent thrombosis are bundled together, and MACE at 30 days have been reported as a surrogate marker of stent thrombosis.
In general, stent thrombosis occurs in 1–2% in different series of patients undergoing percutaneous coronary interventions.30 It is associated with a high rate of morbidity and mortality (20.8% mortality, and 56.6% MI rates at 6 months).31 This degree of morbidity and mortality have led to attempts at characterizing the risk factors for stent thrombosis and hypercoagulability that are outlined in Tables 1, 2 and 3.32–40
In the case presented, the major risk factor for stent thrombosis was hypercoagulability secondary to protease inhibitor therapy. There was no history of hypercoagulability other than HIV disease, nor was there a family history of it. Our patient was diabetic and the final vessel caliber was < 3 mm (postprocedural quantitative coronary angiography revealed that the reference vessel diameter was 2.4 mm). There was TIMI 3 flow postprocedure with good angiographic appearance. IVUS was not utilized, thus it was impossible to determine malapposition or tissue prolapse. Furthermore, the patient was not worked up for any other underlying hypercoagulability. To our knowledge, this is the first stent thrombosis case associated with protease inhibitors in a HIV patient that is accountable for acquired hypercoagulability. Therefore, clinicians should be alert to unprovoked thrombosis episodes as a complication in patients with HIV infection. Optimizing anticoagulant regimen is warranted in patients undergoing PCI in this setting.
Summary
HIV patients are noted to have baseline abnormal coronary artery pathology, arteriopathy/endothelial dysfunction, hypertriglyceridemia and hypercoagulability prior to the availability of protease inhibitors. With the introduction of HAART, particularly protease inhibitors, severe premature coronary artery disease, insulin resistance, fat redistribution syndrome, endothelial dysfunction, hyperlipidemia (metabolic syndrome), hypercoagulability and increased tendency to MI became exacerbating factors for underlying atherosclerotic risk. Appropriate treatment of hyperlipidemia, cardiac risk factor modification, and perhaps careful HAART drug selection, will be helpful in this regard.
We dedicate this article to physicians who help the science to progress.
References
- Quinn TC. The epidemiology of the acquired immunodeficiency syndrome in the 1990’s. Emerg Med Clin North Am 1995;13:1–25.
- Mellors JW, Kingsley LA, Rinaldo CR Jr, et al. Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Ann Int Med 1995;122:573–579.
- Hammer SM, Saag MS, Schechter M, et al. Treatment for adult HIV infection: 2006 recommendations of the international AIDS society-USA panel. JAMA 2006;296:827–843.
- Yunis NA, Stone VE. Cardiac manifestations of AIDS: A review of disease spectrum and clinical management. J Acquir Immune Defic Syndr Hum Retrovirol 1998;18:145–154.
- Cotton P. AIDS giving rise to cardiac problems. JAMA 1990;263:2149.
- Joshi VV, Pawel B, Conner E, et al. Arteriopathy in children with AIDS. Pediatr Pathol 1987;7:261–275.
- Paton P, Tabib A, Loire R, et al. Coronary artery lesions and human immunodeficiency virus infection. Res Virol 1993;144:225–231.
- Soentjens P, Ostyn B, Van Outryve S, et al. Portal vein thrombosis, in a patient with HIV treated with a protease containing regimen. Acta Clinica Belgica 2006;61:24–29.
- George SL, Swindells S, Knudson R, et al. Unexplained thrombosis in HIV-infected patients receiving protease inhibitors: Report of seven cases. Am J Med 1999;107:624–630.
- 10. Shen YM, Frenkel EP. Thrombosis and a hypercoagulable state in HIV infected patients. Clin Appl Thromb Hem 2004;10:277–280.
- Feffer SE, Fox RL, Orsen MM, et al. Thrombotic tendencies and correlation with clinical status in patients infected with HIV. Southern Med J 1995;88:1126–1130.
- Majluf-Cruz A, Silva-Estrada M, Sanchez-Barboza R, et al. Venous thrombosis among patients with AIDS. Clin Appl Thromb Hem 2004;10:19–25.
- Saif MW, Greenberg B. HIV and thrombosis: A review. AIDS Patient Care STDs 2001;15:15–24.
- Henry K, Melroe H, Huebsch J, et al. Severe premature coronary artery disease with protease inhibitors. Lancet 1998;351:1328.
- Karmochkine M, Raguin G. Severe coronary artery disease in a young HIV-infected man with no cardiovascular risk factor who was treated with Indinavir. AIDS 1998;12:2499.
- Passalaris JD, Sepkowitz KA, Glesby MJ. Coronary artery disease and human immunodeficiency virus infection. Clin Infect Dis 2000;31:787–797.
- Lundgren JD, for the DAD study group. Combination antiretroviral therapy and risk of myocardial infarction. N Engl J Med 2003;349:1993–2003.
- Dotter CT. Transluminally placed coil spring arterial tube grafts: Long-term patency in canine popliteal artery. Invest Radiol 1969;4:329.
- Sigwart U, Puel J, Mirkovitch V, et al. Intravascular stents to prevent occlusion and restenosis after transluminal angioplasty. N Engl J Med 1987;316:701–706.
- Serruys PW, Strauss BH, Beatt KJ, et al. Angiographic follow-up after placement of a self-expanding coronary artery stent. N Engl J Med 1991;324:13.
- Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of balloon expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. BENESTENT study group. N Engl J Med 1994;331:489–495.
- Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary stent placement and balloon angioplasty in the treatment of coronary artery disease: Stent restenosis study investigators. N Engl J Med 1994;331:496–501.
- Shaknovich A, Moses JW, Bailey S, et al. Subacute stenosis in the stent restenosis study (STRESS): Clinical impact and predictive factors. Circulation 1994;90:650A.
- Mak KH, Belli G, Ellis S, et al. Subacute stent thrombosis, evolving issues and current concepts. J Am Coll Cardiol 1996;27:494–503.
- Colombo A, Hall P, Nakamura S, et al. Intracoronary stenting without anticoagulation accomplished with intravascular ultrasound. Circulation 1995;91:676.
- Cutlip DE, Leon MB, Ho KK, et al. Acute and nine month clinical outcomes after “suboptimal” coronary stenting: Results from the Stent Anti-thrombotic Regimen Study (STARS) registry. J Am Coll Cardiol 1999;34:698–706.
- De Servi S, Repetto S, Klugmann S, et al. Stent thrombosis: Incidence and related factors in the RISE Registry (Registro Implanto Stent Endocoronarico). Catheter Cardiovasc Interv 1999;46:13–18.
- Karillon G, Morice M, Benveniste E, et al. Intracoronary stent implantation without ultrasound guidance and with replacement of conventional anticoagulation by antiplatelet therapy: 30-day clinical outcome of the French Multicenter Registry. Circulation 1996;74:1519–1527.
- Schomig A, Neumann FJ, Kastrati A, et al A randomized comparison antiplatelet and anticoagulant therapy after the placement of coronary artery stents. N Engl J Med 1996;334:1084–1089.
- Topol EJ, Serruys PW. Frontiers in interventional cardiology. Circulation 1998;98:1802–1820.
- Cutlip D, Baim DS, Ho KKL, et al. Stent thrombosis in the modern era: A pooled analysis of multicenter coronary stent clinical trials. Circulation 2001;103:1967–1971.
- Schuhlen H, Kastrati A, Dirschinger J, et al. Intracoronary stenting and risk for major adverse cardiac events during the first month. Circulation 1998;98:104–111.
- Moussa I, Mario C, Reimers B, et al. Subacute stent thrombosis in the era of intravascular ultrasound-guided coronary stenting without anticoagulation: Frequency, predictors, and clinical outcome. J Am Coll Cardiol 1997;29:6–12.
- Kerr AJ, Stewart RA, Low CJ, et al. Long stenting in native coronary arteries: Relation between vessel size and outcome. Cathet Cardiovasc Diagn 1998;44:170–174.
- Lansky AJ, Roubin GS, O`Shaugnessy CD, et al. Randomized comparison of GR-II stent and Palmaz-Schatz stent for elective treatment of coronary stenosis. Circulation 2000;102:1364–1368.
- Walter DH, Schachinger V, Elsner M, et al. Platelet glycoprotein IIIa polymorphism and risk of coronary stent thrombosis. Lancet 1997;350:1217–1219.
- Laule M, Cascorbi I, Stangl V, et al. A1/A2 polymorphism of glycoprotein IIIa and association with excess procedural risk for coronary catheter interventions: A case controlled study. Lancet 1999;353:687–688.
- Bauer KA, Rosendaal FR, Heit JA. Hypercoagulability: Too many tests, too much conflicting data. American Society of Hematology Educational Book 2002;353–368.
- Schaefer AI, Levine MN, Konkle BA, et al. Thrombotic disorders: Diagnosis and treatment. American Society of Hematology Educational Book 2003;520–539.
- Weitz JI, Middeldorp S, Geerts W, et al. Thrombophilia and new anticoagulant drugs. In: American Society of Hematology Educational Book 2004, Washington, D.C., pp. 424–438.