

Advancing Value in IPF Management With Nerandomilast
Executive Summary
- Idiopathic pulmonary fibrosis (IPF) is a progressive, fatal lung disease with high clinical and economic burden. Current treatment options suffer from tolerability limitations and lack of understanding regarding long-term disease impact.
- Nerandomilast, a novel PDE4B inhibitor, represents a new mechanism of action and demonstrates sustained reductions in forced vital capacity (FVC) through week 76 in the FIBRONEER-IPF trial.
- Nerandomilast demonstrated a favorable safety and tolerability profile that remained consistent over time, supporting its use in patients intolerant to existing antifibrotics and enabling long-term adherence.
- By slowing disease progression and improving persistence, nerandomilast may reduce downstream health care use, including hospitalizations, acute exacerbation care, oxygen dependence, and end-stage care.
- Nerandomilast can be used as a monotherapy or add-on therapy. This flexibility supports individualized treatment approaches and broader clinical use.
- The FIBRONEER-IPF trial data supports nerandomilast’s value across efficacy, safety, adherence, and potential economic impact.
Introduction
Distinguishing IPF From Other Fibrotic ILDs
Interstitial lung disease (ILD) is a devastating chronic lung disorder characterized by inflammation and/or fibrosis of the alveolar interstitium.1 Disease progression is inevitable, and it is estimated that approximately 26,000 ILD patients die in the US each year.1,2 Within ILD, several distinct subclasses of lung disease exist, including idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF).1 While IPF has distinct diagnostic criteria, PPF refers to a unique disease pattern defined by worsening fibrosis, not initial etiology.1
IPF makes up nearly 30% of all ILD cases, and the average survival rate is 3–5 years from diagnosis.1,2 The underlying cause of IPF is unknown, but genetic and environmental factors are thought to play a role in disease onset and progression.2 Patients with IPF often receive a poor prognosis, further complicated by comorbidities such as hypertension, chronic obstructive pulmonary disease (COPD), diabetes, and emphysema.2 The course of IPF disease progression varies across patients, with some remaining stable for long periods while others experience a quick decline in overall health, leading to death.3 Mortality in IPF has been associated with several clinical factors, including advanced age, impaired diffusion capacity of the lungs for carbon monoxide (DLCO), reduced forced vital capacity (FVC), smoking history, comorbidities, and the occurrence of acute exacerbations.3
For patients with IPF, acute exacerbations are a major predictor of morbidity and mortality. The average survival time following an exacerbation event is 3–4 months.3 Given the severity of IPF disease progression, there remains a critical need for novel disease management strategies and therapeutic options to improve the quality of life for patients living with IPF.
The IPF Treatment Landscape: Limited Options, Persistent Gaps
The current treatment landscape for IPF has remained largely unchanged for the past decade, with 2 antifibrotic agents—nintedanib and pirfenidone—serving as the standard of care since their approvals in 2014.2,4,5 Both therapies have demonstrated the ability to slow the rate of lung function decline and are recommended in clinical guidelines for patients with IPF.6 Nintedanib is a multi-target tyrosine kinase inhibitor that affects pathways involved in fibrosis.4 Approved in 2014 for IPF, nintedanib was granted an additional indication for PPF in 2020 based on the phase 3 INBUILD RCT (PPF), INPULSIS-1 and INPULSIS-2 (IPF) trials.1,4 Also approved in 2014, pirfenidone is a multi-pathway inhibitor that primarily acts on transforming growth factor-beta (TGF-β), signaling to exert antifibrotic and anti-inflammatory effects.2 Pirfenidone is solely indicated for IPF based on the results of the phase 3 CAPACITY and ASCEND trials.2,5
Despite their different mechanisms of action, both nintedanib and pirfenidone provide similar clinical benefits in reducing disease progression but are unable to reverse fibrosis or significantly alter long-term survival outcomes.2 Patient characteristics, comorbidities, and tolerability considerations often guide IPF treatment with these agents. However, the utility of both antifibrotic medications is limited by their safety and tolerability profiles. Both nintedanib and pirfenidone are known to lead to frequent dose reductions, interruptions, or discontinuation in real-world IPF management.7,8 Nintedanib is known to cause gastrointestinal adverse events, particularly diarrhea, as well as liver toxicity.2 Similarly, pirfenidone causes nausea, dermatological adverse reactions, liver toxicity, and central nervous system (CNS)-related events.9 These tolerability challenges can compromise treatment persistence and, consequently, long-term effectiveness.
The Escalating Clinical and Economic Burden of Disease Progression in IPF
 With no new treatment options available in the last 12 years, patients with IPF are left cycling between antifibrotic medications that temporarily slow the disease and come with unavoidable toxicity. In late-stage IPF, lung transplantation remains the only definitive therapy option for advanced disease.1,2 The progressive decline of IPF patients also results in medical and financial consequences.
With no new treatment options available in the last 12 years, patients with IPF are left cycling between antifibrotic medications that temporarily slow the disease and come with unavoidable toxicity. In late-stage IPF, lung transplantation remains the only definitive therapy option for advanced disease.1,2 The progressive decline of IPF patients also results in medical and financial consequences.
Acute exacerbations in IPF are unpredictable, often severe, events characterized by rapid respiratory deterioration, and are associated with high short-term mortality and intensive health care utilization.8 These episodes frequently result in hospitalizations, which represent a major driver of IPF-related costs, and are linked to further functional decline and reduced survival.8 As the disease progresses, many patients require long-term supplemental oxygen therapy to manage chronic hypoxemia, adding to both clinical burden and ongoing health care expenses.1 Together, exacerbations, hospitalizations, and oxygen dependence reflect advanced disease progression and contribute substantially to the overall economic and quality-of-life impact of IPF.
Nerandomilast: A New Standard Emerging in IPF
In October 2025, nerandomilast received FDA approval for the treatment of IPF in adult patients, marking the first new antifibrotic approved for IPF in nearly 12 years.10 Nerandomilast is a novel, orally administered phosphodiesterase (PDE) 4B inhibitor designed to target key inflammatory and fibrotic pathways implicated in the progression of IPF.11 By inhibiting PDE4B, nerandomilast increases intracellular cyclic adenosine monophosphate (cAMP) levels, leading to suppression of downstream pro-inflammatory and profibrotic signaling cascades.12 This results in reduced production of cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1β, as well as decreased chemokine release and immune cell recruitment.13 By suppressing these signaling pathways, nerandomilast directly modulates the inflammatory environment that contributes to fibrosis development and progression in IPF.12
Nerandomilast exerts direct antifibrotic activity at the cellular level. Preclinical and translational studies indicate that PDE4B inhibition can inhibit fibroblast proliferation, reduce the differentiation of fibroblasts into myofibroblasts, and decrease extracellular matrix deposition.12,14 Each of these cellular processes are known drivers of irreversible lung scarring. In addition, nerandomilast interferes with TGF-β-mediated signaling, a central pathway in fibrogenesis.12 By targeting multiple points in the fibrotic signaling cascade, nerandomilast offers a mechanistically distinct approach compared to nintedanib and pirfenidone by broadly modulating both inflammatory and fibrotic drivers of disease.
 The rationale for targeting PDE4 in IPF is grounded in its role as the dominant cAMP-degrading enzyme in immune and inflammatory cell populations.12,13 Elevated PDE4 activity contributes to reduced intracellular cAMP levels, thereby promoting inflammatory signaling and fibrotic responses. Inhibiting PDE4 restores cAMP-mediated regulatory pathways, directly impacting the cellular processes that underlie fibrotic progression.14 Importantly, nerandomilast is selective for the PDE4B isoform, which differentiates it from earlier non-selective PDE4 inhibitors that also target PDE4D.14 The non-selective inhibitors have historically been associated with increased gastrointestinal and CNS adverse reactions, limiting their clinical utility. By preferentially targeting PDE4B, nerandomilast maintains therapeutic activity while improving tolerability, supporting its potential for long-term use in patients with IPF.12
The rationale for targeting PDE4 in IPF is grounded in its role as the dominant cAMP-degrading enzyme in immune and inflammatory cell populations.12,13 Elevated PDE4 activity contributes to reduced intracellular cAMP levels, thereby promoting inflammatory signaling and fibrotic responses. Inhibiting PDE4 restores cAMP-mediated regulatory pathways, directly impacting the cellular processes that underlie fibrotic progression.14 Importantly, nerandomilast is selective for the PDE4B isoform, which differentiates it from earlier non-selective PDE4 inhibitors that also target PDE4D.14 The non-selective inhibitors have historically been associated with increased gastrointestinal and CNS adverse reactions, limiting their clinical utility. By preferentially targeting PDE4B, nerandomilast maintains therapeutic activity while improving tolerability, supporting its potential for long-term use in patients with IPF.12
FIBRONEER-IPF: Pivotal Evidence in IPF
The phase 3 FIBRONEER-IPF trial was designed to address two critical gaps in the management of IPF: the need for therapies that meaningfully slow disease progression and the need for improved tolerability to support long-term treatment adherence.7 Specifically, the study aimed to evaluate whether nerandomilast could improve clinical outcomes, as measured by preservation of lung function, while offering a safety and tolerability profile that limits dose modifications and discontinuations commonly observed with nintedanib and pirfenidone.7
FIBRONEER-IPF was a global, randomized, double-blind, placebo-controlled trial that enrolled a total of 1,177 patients, reflecting one of the more robust phase 3 datasets in IPF.7 The study included patients receiving background antifibrotic therapy with nintedanib and pirfenidone, reflecting a real-world-relevant clinical population, as well as patients receiving only nerandomilast.7 Patients were randomized to receive either placebo, nerandomilast 9 mg, or nerandomilast 18 mg—each twice daily.7 Patients were stratified by background therapy and assessed at regular intervals through week 52.7 The primary endpoint was the absolute change from baseline in FVC at week 52, a validated surrogate for disease progression and mortality risk in IPF.7,15 Nerandomilast met the primary endpoint at both dose levels, demonstrating a statistically significant reduction in FVC decline compared to placebo.7
 The trial monitored adverse events historically associated with PDE4 inhibition, including gastrointestinal toxicity, psychiatric events, and vasculitis.7 Overall, nerandomilast demonstrated a favorable safety profile, with comparable adverse event-related discontinuation rates to placebo.7 The pivotal trial results from FIBRONEER-IPF established nerandomilast as the first novel antifibrotic therapy approved for IPF in over a decade and marked it as a valuable addition to the IPF treatment paradigm, with the potential to improve both clinical and economic outcomes through enhanced disease control and treatment persistence.
The trial monitored adverse events historically associated with PDE4 inhibition, including gastrointestinal toxicity, psychiatric events, and vasculitis.7 Overall, nerandomilast demonstrated a favorable safety profile, with comparable adverse event-related discontinuation rates to placebo.7 The pivotal trial results from FIBRONEER-IPF established nerandomilast as the first novel antifibrotic therapy approved for IPF in over a decade and marked it as a valuable addition to the IPF treatment paradigm, with the potential to improve both clinical and economic outcomes through enhanced disease control and treatment persistence.
FIBRONEER-IPF: Extended Follow-Up Results
Long-Term Efficacy Through Week 76
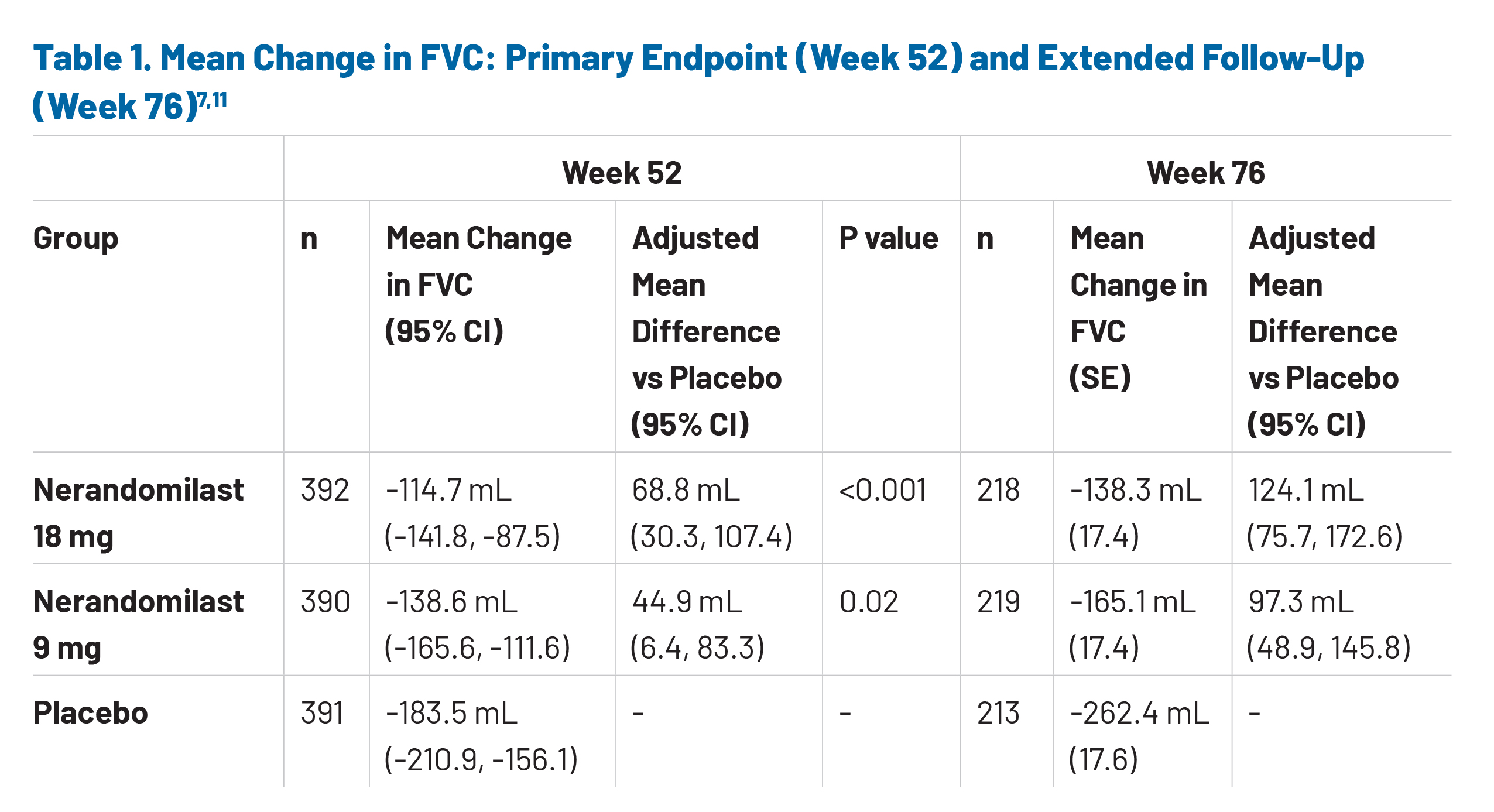
 Extended follow-up data from the FIBRONEER-IPF trial provide key insights into the durability of nerandomilast and its potential long-term value in the IPF market. Notably, mean treatment exposure was similar across all groups, averaging approximately 14.7–14.9 months for placebo and both nerandomilast doses (9 mg and 18 mg), regardless of treatment interruptions.11 This consistency suggests good tolerability and sustained adherence. Collectively, 85.2% of enrolled trial participants completed the observation period through the final database lock.11 Regarding the efficacy of nerandomilast, the reduction in FVC decline observed at the primary endpoint (week 52) continued through week 76, indicating sustained disease-modifying activity over time (Table 1).11 These data demonstrate a clinically meaningful reduction in lung function decline with persistent nerandomilast use.11 The continued separation of FVC trajectories reinforces the role of nerandomilast in slowing disease progression beyond the initial treatment period, which is particularly relevant in a chronic, progressive condition such as IPF, where long-term preservation of lung function is closely tied to patient quality of life and overall health outcomes.11,16
Extended follow-up data from the FIBRONEER-IPF trial provide key insights into the durability of nerandomilast and its potential long-term value in the IPF market. Notably, mean treatment exposure was similar across all groups, averaging approximately 14.7–14.9 months for placebo and both nerandomilast doses (9 mg and 18 mg), regardless of treatment interruptions.11 This consistency suggests good tolerability and sustained adherence. Collectively, 85.2% of enrolled trial participants completed the observation period through the final database lock.11 Regarding the efficacy of nerandomilast, the reduction in FVC decline observed at the primary endpoint (week 52) continued through week 76, indicating sustained disease-modifying activity over time (Table 1).11 These data demonstrate a clinically meaningful reduction in lung function decline with persistent nerandomilast use.11 The continued separation of FVC trajectories reinforces the role of nerandomilast in slowing disease progression beyond the initial treatment period, which is particularly relevant in a chronic, progressive condition such as IPF, where long-term preservation of lung function is closely tied to patient quality of life and overall health outcomes.11,16
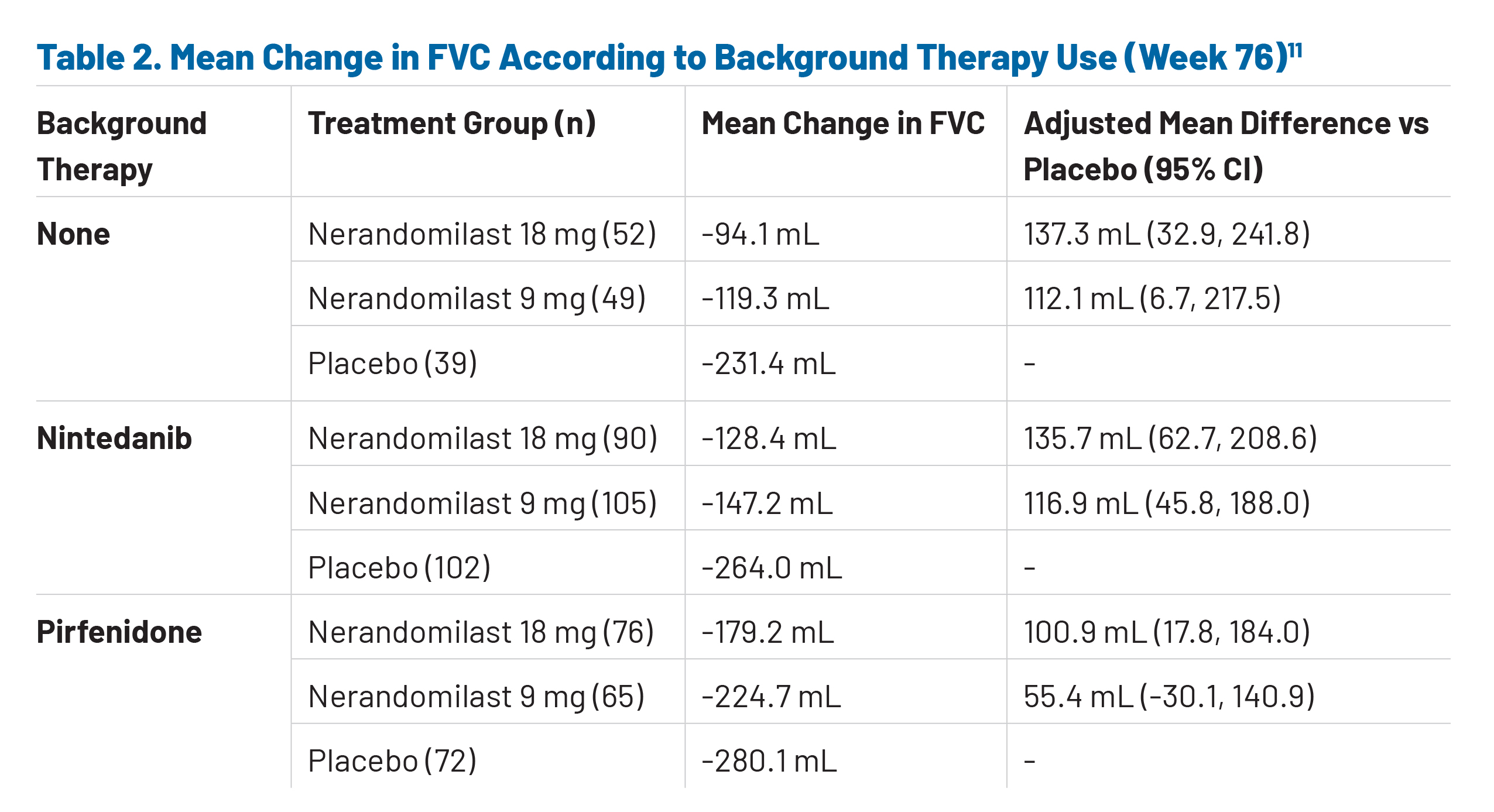
Subgroup analyses from the extended follow-up period provide important context regarding treatment effects in patients receiving background antifibrotic therapy. Among patients treated with background nintedanib, the placebo group experienced a substantial decline in lung function, with a mean FVC change of approximately -264.0 mL at week 76, underscoring the progressive nature of the disease despite standard-of-care therapy (Table 2).11 This finding highlights two key considerations: first, that nintedanib alone does not fully halt disease progression, leaving a meaningful residual unmet need; and second, that the study population represents a clinically advanced and high-risk cohort. Notably, both doses of nerandomilast demonstrated attenuation of FVC decline in the nintedanib subgroup, indicating that nerandomilast retains efficacy even in patients with ongoing progression on background therapy (Table 2).11
In contrast, results in patients receiving background pirfenidone showed a less pronounced treatment effect, with smaller differences between nerandomilast and placebo groups (Table 2).11 Importantly, concomitant use of pirfenidone has been associated with an approximately 50% reduction in plasma concentrations of nerandomilast, which may have contributed to the observed reduction in efficacy.11 The pharmacokinetic relationship between pirfenidone and nerandomilast must be carefully considered when interpreting data from this treatment subgroup
 Time-to-event analyses further contextualize these findings. For all secondary endpoints, the number of events increased between the first and final database locks; however, the increase was smaller among patients receiving nerandomilast than among those receiving placebo.11 By week 76, the proportion of patients experiencing acute exacerbations or death was numerically lower with nerandomilast compared with placebo.11 Similarly, respiratory hospitalization or death occurred in a greater percentage of placebo-treated patients versus patients receiving nerandomilast (Table 3).11 While these differences are not statistically significant, they demonstrate consistent directional trends across clinically meaningful outcomes. Importantly, subgroup analyses suggest that the magnitude of benefit may vary by background therapy, with signals of greater efficacy in patients not receiving concomitant antifibrotics—an observation that should be further studied to inform future nerandomilast positioning and treatment sequencing strategies.11
Time-to-event analyses further contextualize these findings. For all secondary endpoints, the number of events increased between the first and final database locks; however, the increase was smaller among patients receiving nerandomilast than among those receiving placebo.11 By week 76, the proportion of patients experiencing acute exacerbations or death was numerically lower with nerandomilast compared with placebo.11 Similarly, respiratory hospitalization or death occurred in a greater percentage of placebo-treated patients versus patients receiving nerandomilast (Table 3).11 While these differences are not statistically significant, they demonstrate consistent directional trends across clinically meaningful outcomes. Importantly, subgroup analyses suggest that the magnitude of benefit may vary by background therapy, with signals of greater efficacy in patients not receiving concomitant antifibrotics—an observation that should be further studied to inform future nerandomilast positioning and treatment sequencing strategies.11

Mortality outcomes, although limited by event rates and study duration, offer additional context for understanding the potential clinical impact of nerandomilast in IPF. Death occurred in 42 patients (10.7%) in the placebo group compared with 36 (9.2%) and 26 (6.6%) patients in the nerandomilast 9 mg and 18 mg groups, respectively, with the higher dose associated with a numerically lower risk of death.11 Respiratory-related deaths occurred in 6.4% of placebo-treated patients and in 5.9% and 4.1% of patients receiving 9 mg and 18 mg nerandomilast, respectively.11 Consistent trends were also observed for composite measures of disease progression, including absolute decline in FVC ≥10% or death (38.9% with placebo vs 33.7% and 29.6% with 9 mg and 18 mg nerandomilast, respectively) and decline in DLCO ≥15% or death (22.9% placebo vs 20.9% and 19.9% with 9 mg and 18 mg nerandomilast, respectively).11 These endpoints are widely recognized as clinically meaningful markers of progression and are associated with increased risk of hospitalization, resource utilization, and mortality.
At the time of the week 52 primary endpoint analysis, follow-up duration was insufficient to determine the effect of nerandomilast on long-term mortality.7 The time-to-event endpoints assessed during the extended follow-up period reveal promising numerical trends but were not tested for statistical significance, underscoring the need for longer-term studies on the effect of nerandomilast on acute exacerbation, hospitalization, and death.11
Long-Term Safety Through Week 76
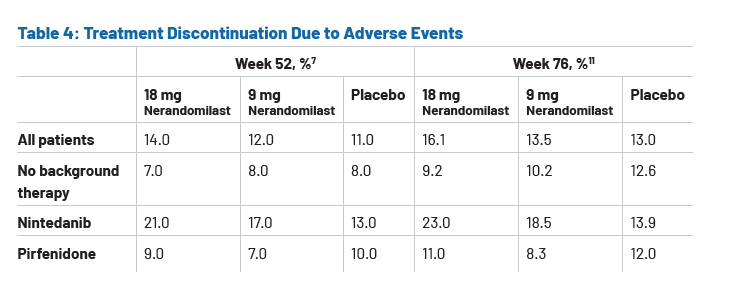
The safety profile observed over the extended follow-up period remained consistent with earlier findings, supporting the long-term tolerability of nerandomilast.11 Discontinuation due to adverse events occurred at similar rates across treatment arms for patients not receiving background therapy.11 However, higher rates of treatment discontinuation due to adverse events were reported for patients receiving nintedanib.11 (Table 4) Diarrhea was the most frequently reported adverse event with nerandomilast, with higher incidence in patients receiving background therapy with nintedanib than in those with no background therapy.11 Incidence rates with background therapy were 31.2% vs 8.0% for placebo, 50.5% vs 17.0% for 9 mg nerandomilast, and 62.4% vs 27.6% for 18 mg nerandomilast.11 However, these events were generally mild to moderate (Common Terminology Criteria for Adverse Events (CTCAE) grade 1 or 2) and infrequently led to discontinuation.11
 Serious adverse events, including psychiatric events, vasculitis, and drug-induced liver injury were balanced across treatment groups, with no new safety signals identified.11 These findings support a favorable risk-benefit profile and suggest that nerandomilast may enable sustained treatment adherence, a key driver of real-world effectiveness in IPF management.11
Serious adverse events, including psychiatric events, vasculitis, and drug-induced liver injury were balanced across treatment groups, with no new safety signals identified.11 These findings support a favorable risk-benefit profile and suggest that nerandomilast may enable sustained treatment adherence, a key driver of real-world effectiveness in IPF management.11
Discussion
Implications for Clinical Practice
The extended findings from the FIBRONEER-IPF trial reinforce nerandomilast as a clinically meaningful addition to the IPF treatment arsenal with sustained disease-modifying effects beyond the initial 52-week analysis. The measured difference in FVC between nerandomilast and placebo began early in the study, with a significant difference at week 52.11 The greatest separation was seen at 76 weeks, indicating sustained attenuation of functional decline over time.11 For clinicians, this durability matters, as IPF is a chronic, relentlessly progressive disease. Preserving lung function over longer periods of time may translate into slower clinical deterioration, prolonged independence, and delayed escalation to oxygen supplementation, hospitalization, or end-stage supportive care.17
Additionally, subgroup findings have potential implications for treatment sequencing of nerandomilast. Some of the most favorable directional effects were observed in patients not receiving background antifibrotic therapy, suggesting that nerandomilast may warrant consideration earlier in the treatment pathway or in selected patients for whom currently available agents are poorly tolerated.11 Equally important is that the safety profile remained favorable with longer exposure.11 For patients who require long-term management, this tolerability profile may support improved treatment persistence and, in turn, improved real-world effectiveness than is often achieved when therapy is interrupted, dose-reduced, or discontinued.3 Collectively, the FIBRONEER-IPF results support the role of nerandomilast in improving long-term disease control and offer important considerations for optimizing treatment strategies in patients with IPF.
Economic Impact: Payer Considerations
The implications of the longer-term results from FIBRONEER-IPF extend beyond clinical differentiation. Acute exacerbations and respiratory hospitalizations in IPF are unpredictable, severe, and resource-intensive, often accelerating decline and increasing mortality risk.3 A therapy that slows progression may help defer these downstream events. Because FVC decline is closely linked to mortality risk and worsening disease, sustained preservation of lung function may reasonably be viewed as relevant to both long-term economic burden and clinical benefit over time.17 The tolerability profile of nerandomilast may further improve adherence and persistence, reducing the hidden costs associated with treatment failure, switching, or unmanaged progression.17
 FDA approval of nerandomilast introduces a novel mechanism of action through selective PDE4B inhibition, expanding therapeutic options in a disease area that has seen limited innovation over the past decade. By combining sustained efficacy, manageable toxicity, and potential flexibility as both monotherapy and add-on therapy, nerandomilast is poised to be a valuable addition to the IPF treatment landscape.11 Nerandomilast offers payers a differentiated treatment option with potential to improve long-term disease control and treatment persistence.11,17
FDA approval of nerandomilast introduces a novel mechanism of action through selective PDE4B inhibition, expanding therapeutic options in a disease area that has seen limited innovation over the past decade. By combining sustained efficacy, manageable toxicity, and potential flexibility as both monotherapy and add-on therapy, nerandomilast is poised to be a valuable addition to the IPF treatment landscape.11 Nerandomilast offers payers a differentiated treatment option with potential to improve long-term disease control and treatment persistence.11,17
Future Directions
While the FIBRONEER-IPF trial establishes a strong foundation for nerandomilast in the treatment of IPF, several areas warrant further investigation to fully determine its long-term clinical and real-world value. Longer-term evidence is needed to confirm the impact of nerandomilast on high-priority outcomes for patients, clinicians, and caregivers.11 Although promising directional trends were observed in the extended follow-up period, longer treatment and observation periods as well as real-world data will be critical to validate these effects.
Evaluating nerandomilast in real-world populations will also be essential, particularly with respect to treatment adherence and discontinuation rates relative to current standard-of-care antifibrotics. Given the favorable tolerability profile observed, understanding how nerandomilast-based therapy translates into long-term use in routine practice will help clarify its potential to improve disease progression and health care resource utilization.
Additional research should focus on defining the optimal role of nerandomilast within the evolving IPF treatment paradigm. This includes evaluating its positioning as an early-line versus later-line therapy, as well as its use as an alternative or add-on to nintedanib and pirfenidone. Further exploration in treatment-naïve patients and deeper assessment of drug-drug interactions will also be critical to guide clinical decision-making.11 Translating clinical promise into payer confidence relies on addressing these priorities, enabling informed decisions on access, positioning, and long-term value in IPF management.
Conclusion
The FIBRONEER-IPF trial establishes PDE4B inhibition as a validated anti-inflammatory and antifibrotic approach in IPF, demonstrating that nerandomilast provides sustained, clinically meaningful reductions in lung function decline.11 At week 76, patients treated with nerandomilast experienced substantially less decline in FVC compared with placebo (-165.1 mL and -138.3 mL for 9 mg and 18 mg respectively, vs -262.4 mL for placebo).11 The measured difference in FVC between treatment groups began early in the study, with significant difference at week 52 continuing through week 76, indicating durable and consistent response to nerandomilast.11
Directional improvements were also observed across key clinical outcomes, including lower proportions of patients experiencing acute exacerbations or death (15.6% and 13.8% for 9 mg and 18 mg nerandomilast, respectively, vs 17.3% for placebo) and a numerically reduced risk of death for patients receiving 18 mg nerandomilast [HR (95% CI) 0.95 (0.61, 1.49) for 9 mg and 0.66 (0.41, 1.08) for 18 mg nerandomilast].11 These findings support the role of nerandomilast as a disease-modifying therapy with the potential to impact outcomes closely linked to survival and health care utilization.
The safety and tolerability profile of nerandomilast—characterized by low rates of treatment discontinuation and no new safety signals—distinguishes it from existing therapies such as nintedanib and pirfenidone.11 This improved tolerability has the potential to support increased time spent on treatment, fewer dose modifications, and broader patient eligibility, including patients unable to tolerate nintedanib or pirfenidone.11 As a result, nerandomilast may enable more consistent disease control in real-world settings, where treatment persistence is a key determinant of effectiveness and a major challenge in IPF management.
The evidence from FIBRONEER-IPF positions nerandomilast as a meaningful advancement in the IPF treatment landscape. By combining a novel mechanism of action, sustained efficacy, and a manageable safety profile, nerandomilast offers the potential to improve long-term disease control and patient outcomes. These attributes may ultimately translate to reduced downstream health care burdens by delaying disease progression, preserving functional status, and improving survival in patients living with IPF.
References
1. Maher TM. Interstitial lung disease: a review. JAMA. 2024;331(19):1655-1665. doi:10.1001/jama.2024.3669.
2. Glass DS, Grossfeld D, Renna HA, et al. Idiopathic pulmonary fibrosis: current and future treatment. Clin Respir J. 2022;16:84-96. doi:10.1111/crj.13466.
3. Onyilmaz T, Baris SA, Ozturk B, et al. Evaluation of factors affecting mortality in patients with idiopathic pulmonary fibrosis: a 10-year single-center experience. Diagnostics (Basel). 2026;16(1):74. doi:10.3390/diagnostics16010074.
4. OFEV® (nintedanib) capsules, for oral use. Prescribing information. Boehringer Ingelheim Pharmaceuticals, Inc. 2022. Accessed April 13, 2026. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/205832Orig1s016lbl.pdf
5. ESBRIET® (pirfenidone) capsules, for oral use. Prescribing Information. InterMune, Inc. 2014. Accessed April 13, 2026. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022535s000lbl.pdf
6. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3-e19. doi:10.1164/rccm.201506-1063ST.
7. Richeldi L, Azuma A, Cottin V, et al. Nerandomilast in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2025. doi:10.1056/NEJMoa2414108.
8. Strykowski R, Adegunsoye A. Idiopathic pulmonary fibrosis and progressive pulmonary fibrosis. Immunol Allergy Clin North Am. 2023;43(2):209-228. doi:10.1016/j.iac.2023.01.010.
9. Gulati S, Luckhardt TR. Updated evaluation of the safety, efficacy and tolerability of pirfenidone in the treatment of idiopathic pulmonary fibrosis. Drug Healthc Patient Saf. 2020;12:85-94. doi:10.2147/DHPS.S224007.
10. U.S. Food and Drug Administration. FDA approves drug to treat idiopathic pulmonary fibrosis. Published October 7, 2025. Accessed April 13, 2026. https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-drug-treat-idiopathic-pulmonary-fibrosis
11. Oldham JM, Azuma A, Kreuter M, et al. Nerandomilast in idiopathic pulmonary fibrosis: data from the whole follow-up period of the FIBRONEER-IPF trial. Am J Respir Crit Care Med. Published online February 21, 2026. doi:10.1093/ajrccm/aamag058.
12. Reininger D, Wolf F, Mayr CH, et al. Insights into the cellular and molecular mechanisms behind the antifibrotic effects of nerandomilast. Am J Respir Cell Mol Biol. 2025;73(5):700-712. doi:10.1165/rcmb.2024-0614OC.
13. Liu Y, Hu Y, Se-rigeleng, Yang Z, et al. Nerandomilast attenuates idiopathic inflammatory myopathy–associated interstitial lung disease via inhibiting proliferation and differentiation of B cells. Front Immunol. 2026;17:1771007. doi:10.3389/fimmu.2026.1771007.
14. Reininger D, Fundel-Clemens K, Mayr CH, Wollin L, Laemmle B, Quast K, Nickolaus P, Herrmann FE. PDE4B inhibition by nerandomilast: effects on lung fibrosis and transcriptome in fibrotic rats and on biomarkers in human lung epithelial cells. Br J Pharmacol. 2024;181(23):4766-4781. doi:10.1111/bph.17303.
15. Maher TM, Bendstrup E, Voss F, et al. Decline in forced vital capacity as a surrogate for mortality in patients with pulmonary fibrosis. Respirology. 2023;28:1147-1153. doi:10.1111/resp.14579.
16. Diamantopoulos A, Wright E, Vlahopoulou K, Cornic L, Schoof N, Maher TM. The burden of illness of idiopathic pulmonary fibrosis: a comprehensive evidence review. Pharmacoeconomics. 2018;36(7):779-807. doi:10.1007/s40273-018-0631-8.
17. Dempsey TM, Thao V, Moriarty JP, Borah BJ, Limper AH. Cost-effectiveness of the anti-fibrotics for the treatment of idiopathic pulmonary fibrosis in the United States. BMC Pulm Med. 2022;22:18. doi:10.1186/s12890-021-01811-0.
© 2026 HMP Global. All Rights Reserved.
Any views and opinions expressed are those of the author(s) and/or participants and do not necessarily reflect the views, policy, or position of First Report Managed Care or HMP Global, their employees, and affiliates.



